
核心提示:一项新发表的基因突变检测方法(SnpC),其突破在于重新定义了遗传多样性的度量层级。传统方法将多样性直接定义在群体;新方法则先将定义下沉到个体——因为突变首先发生在那里,再上浮与群体多样性整合。承认个体异质性,不等于否认群体多样性。两者并非对立,而是层次不同。
中科院昆明动物研究所马占山研究员、张亚平院士研究团队近日在《BMC Bioinformatics》上发表了名为SnpC的计算工具,用于识别人群中携带独特SNP或SNP显著富集的基因。该方法在千人基因组计划的五个主要人群(非洲、欧洲、东亚、南亚、美洲)中,鉴定出数千个突变模式具有人群特异性的基因。这套工具的方法学根基,要追溯到同一团队五年前的一项工作。
传统方法:多样性直接定义在群体
2020年,该团队在《科学报告》上发表论文,指出了一个长期被忽视的方法论事实。
经典的核苷酸多样性等指标,其数学定义直接落在群体层面。它们度量的是一群人内部任意两条序列的平均差异。数据固然来自个体的血液或唾液样本,但度量本身没有“个体版”——不存在“个体核苷酸多样性”这个概念。度量的基本单位是群体,不是人。
这并非技术疏忽,而是学科范式使然。群体遗传学自诞生之初,关注的就是群体层面的演化力量——突变、漂变、迁徙、选择。个体差异被视为样本误差,而非度量的对象。
但这种范式有一个盲区:遗传变异首先发生在个体基因组上。 突变发生于某一个精细胞、某一个卵细胞、某一个受精卵。群体层面的差异,不过是无数个体事件的统计投影。将度量直接定义在群体,等于跳过了变异实际发生的层级。
第一步:将定义下沉到个体
2020年的论文完成了第一步:把度量的定义域从群体下移到个体。借用生态学中基于Rényi熵的Hill数,该研究定义了个体层面的SNP多样性——Alpha多样性(单一个体基因组内部的突变异质性)、Beta多样性(两个个体之间的差异)、Gamma多样性(群体内所有个体的总多样性)。这套框架让研究者第一次能够像测量一棵树的生长状况那样,精确量化单一个体基因组的突变图谱。
这一步的意义,哲学层面大于技术层面:它承认了个体是变异的真正承载者,度量应该从这个层级开始。该研究在技术上提供了“抓手”——个体Alpha、Beta、Gamma多样性指标让研究者第一次能够量化单一个体基因组的突变图谱。但哲学的转向需要方法的跟进。如何将个体层面的度量用于人群比较?如何在全基因组尺度上判断哪些基因的突变模式具有人群特异性?这些问题的回答,依赖于方法上的进一步突破。
这正是SnpC完成的工作。
第二步:上浮并与群体多样性整合
有了个体层面的度量,新的问题出现了:怎么用它来比较人群?这正是SnpC完成的工作。它定义了一个名为“基因-SNP异质性”(GSH)的指标,将个体层面的SNP信息聚合到群体层面,综合了两类信息:该人群中有多大比例的个体在目标基因上携带SNP,以及该基因的总SNP量在各人群之间如何分配。
这一步的关键在于整合而非替代。SnpC没有抛弃传统的群体比较框架,而是在个体度量的基础上重新构建了群体比较的逻辑。最终输出的仍然是人群之间的差异,但这个差异的底层是个体图谱的聚合,而非直接对群体求平均。
计算上的硬骨头与实用的近路
接下来的统计检验面临一个计算上的硬骨头。要精确判断观测到的GSH差异是否显著,需要穷尽所有可能的个体分组方式,构建精确的零分布。仅以非洲和欧洲各100个个体为例,可能的分组方式约为 9 × 10⁵⁸ × 9 × 10⁵⁸ = 81 × 10¹¹⁶ 种,可能超过了可观测宇宙的原子总数 (约 1080 的规模)。这是一个NP完全问题,精确求解在计算上不可行。
面对这类问题,理论计算机科学给出的出路是:不求精确解,求近似解。但近似不是随便猜——需要一种算法,能够在可控时间内给出一个统计上可靠、理论上可辩护的答案。这正是SnpC的核心进展。 它采用的策略是蒙特卡洛置换检验:随机打乱人群标签1000次,每次重新计算GSH差异,用这1000次的结果拼出一个近似的零分布,再据此计算P值。这种做法在统计学上有严格的理论保证——估计是无偏的,精度随抽样次数增加而提高。1000次抽样听起来不多,但足以区分真实信号与随机噪声,且计算量固定、完全可控。
这好比物流公司面对旅行商问题:100个城市的绝对最短路线,穷举法需要计算的时间比宇宙年龄还长。但没有人会等那个最优解。工程师开发出了贪心算法、遗传算法、AI神经网络等一系列启发式算法,能在几秒内算出一个“够好”的路线——虽然不是数学上完美的,但足以让货车按时发车、成本控制在合理范围内。
SnpC做的正是同一类事情。它的贡献不在于宣称“精确解不可能”——这早就是教科书常识;而在于为这个具体的遗传学问题,设计出了那个“够好”的算法。 GSH指标将个体层面的复杂突变分布压缩为一个可计算的标量;置换检验框架则提供了统计显著性的近似推断。两者结合,使得原本在计算上不可行的精确检验,变成了一件研究者用普通计算机就能完成的工作。
这就是从“问题无解”到“问题可解”的跨越。不是理论上的可解——NP完全的帽子还戴着——而是实践上的可解。对每天面对真实数据的遗传学家而言,后者才是真正重要的。
方法发现了什么?
应用于五个主要人群的数据,SnpC给出了清晰的信号,例如:
非洲人群遗传多样性最高。 非洲群体中检出的独特SNP和富集SNP数量远超其他人群。一个非洲个体的基因组中,有些SNP模式在数万年前迁出非洲的后裔中已完全消失。这一结论验证了走出非洲的瓶颈模型,但SnpC的价值在于提供了基因级精度的目录:它告诉你,具体是哪些基因携带了这些人群特有的变异。
嗅觉基因分化显著。 控制嗅觉的基因是人群体间差异最大的基因类别之一。不同人群感知特定气味的能力差异明显,这可能反映了对不同环境和饮食的长期适应。
局限与意义
对“独特基因”的判定需要谨慎。一个在500人欧洲样本中未检出的SNP,不等于整个欧洲人群都不存在。样本量是硬约束,稀有变异容易被遗漏。因此,确定该方法的“最小可靠”样本数,是进一步研究需要解决的课题。
另一个重要的扩展方向,是将该方法从现有的SNP检测,延伸至其他类型的遗传变异,如插入、缺失、拷贝数变异等。作者在论文中也指出,SnpC的框架理论上适用于各类结构变异,但技术实现仍需后续工作来验证。
回看这条方法学链条,逻辑是清晰的:传统方法将多样性直接定义在群体;2020年的论文把定义下沉到个体;SnpC再将个体度量上浮,与群体整合。下沉不是目的,整合才是。最终回答的仍是人群比较的问题,但路径变了——先量树,再测林。
要见森林,但不能忘记树木。对于精准医学,这句话有切实的意义。患者不是群体,其基因组是一幅独一无二的变异图谱。只有从个体出发再回到群体的方法,才可能既看见森林的结构,又不丢失每一棵树的细节。同时,新方法为更好地利用群体信息指导个体层面的诊治,提供了更为科学的工具。
这个“森林与树木”的类比,在生态学中或许有它的限度——森林经营不必也不可能照看每一棵树。但在医学中,照看每一个具体的人,不是技术可行性问题,而是伦理应然问题。新方法的意义正在于此:它为这个应然提供了实然的工具。
论文来源:
2026 SnpC Study
Ma ZS, Li L, Zhang YP. A new SNP comparison (SnpC) method for detecting unique/enriched gene-SNP variants and comparing population gene mutation diversity. BMC Bioinformatics. 2026. DOI: 10.1186/s12859-026-06381-8
2020 Foundational Paper
Ma ZS, Li LW, Zhang YP. Defining individual-level genetic diversity and similarity profiles. Scientific Reports. 2020;10(1):5805. DOI: 10.1038/s41598-020-62362-8
Institutional press release (2020):Kunming Institute of Zoology, Chinese Academy of Sciences. "Scientists Propose New Concept 'Individual-Level Genetic Diversity'." May 2020.
http://english.kiz.cas.cn//ns/rp/202005/t20200527_237811.html
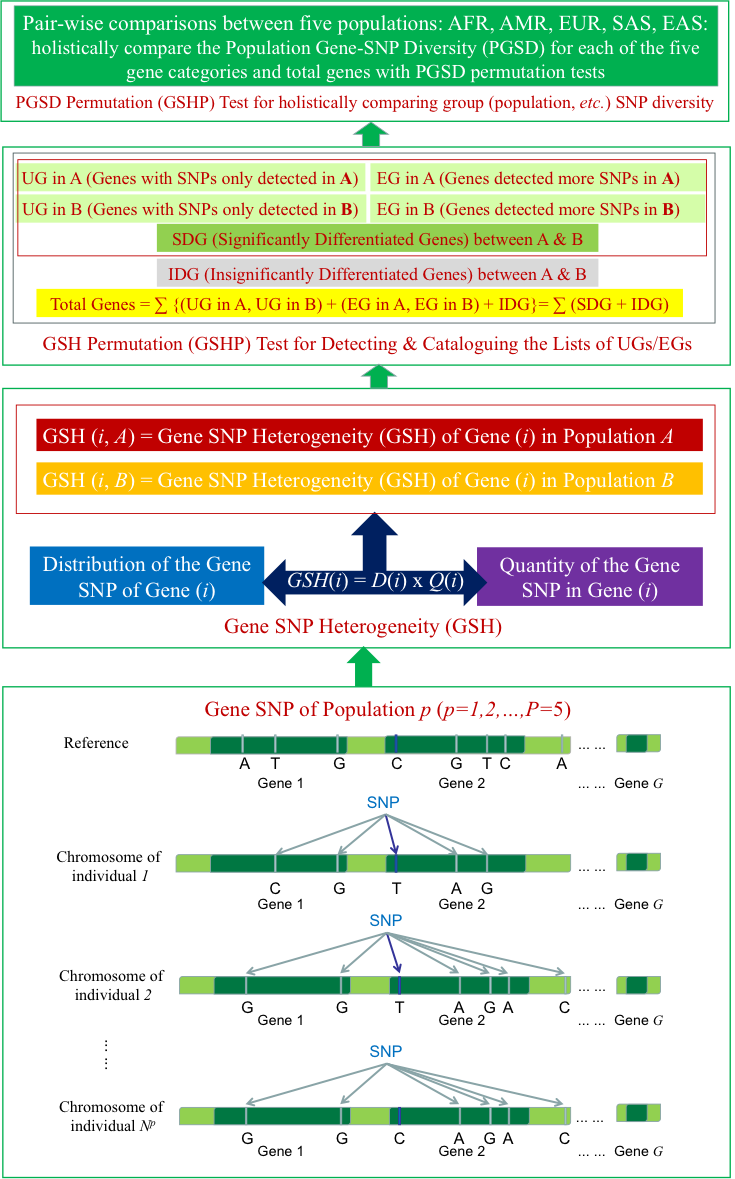
用于检测独特和富集的基因变异的SNP比较(SnpC)框架
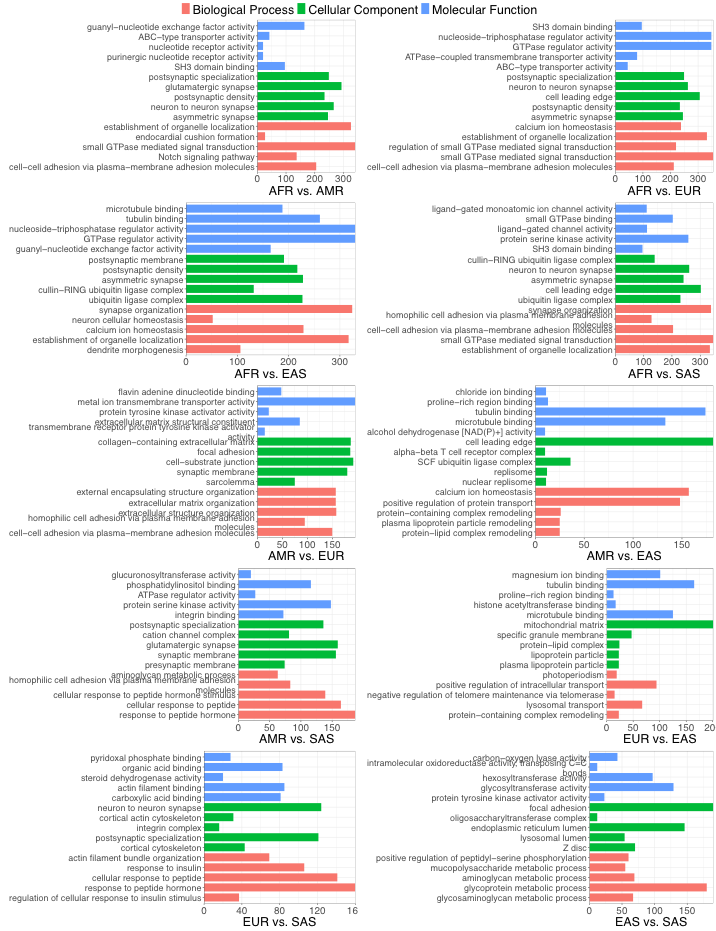
5个种群的10组两两比较,后一群体中下调基因的GO功能
