| Nucleic Acids Res | 识别多状态数据中共有和特异的生物模式的新方法 |
| 2019-06-10 | 作者: 张世华课题组 | 来源: 数学院 【打印】 |
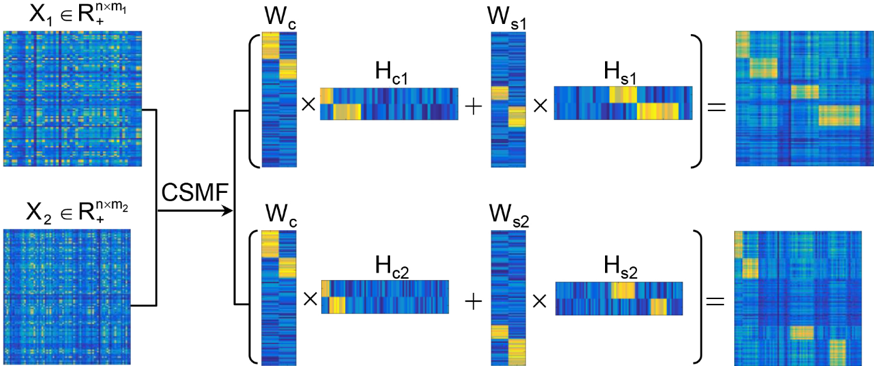
随着高通量测序技术(例如ChIP-seq,RNA-seq和single cell RNA-seq)的发展,逐渐产生并积累了大量的不同条件且相互关联(如不同癌症、不同细胞系、不同时刻)的基因组数据。这为通过大规模数据分析和数据比较,来揭示不同关联条件下存在的共性和特异性提供了机会。整合分析与差异分析是两个常用的数据分析的范式。然而,通常的整合方法忽略了差异的模式,而差异分析方法通常不能识别差异部分的组合模式,一些识别组合模式的方法则要求数据的维数是匹配的。为此,该研究提出了一个基于非负矩阵分解技术,同时识别共有和特异组合模式的强大模型CSMF(Common and Specific patterns via Matrix Factorization)。
图(一)CSMF模型的示意图。 该模型被用于四种生物情景数据(包括不同细胞系的ChIP-seq数据、不同癌症的RNA-seq数据、三种不同的乳腺癌亚型的RNA-seq数据和不同时间点的胚胎干细胞分化的单细胞RNA-seq数据)。通过对这四组不同生物场景下的数据的分析表明,CSMF是一个用来解释相互关联数据中隐藏的组合模式的有效方法。具体来说,应用到ChIP-seq数据时,CSMF可以揭示不同细胞系共有和特异的组蛋白修饰的组合模式以及对应的位点信息。当被应用于不同癌症的基因表达数据时,CSMF可以揭示不同癌症分子机制的异同。进一步,CSMF可以揭示同一种癌症不同亚型之间微小的差异。CSMF还可以应用到单细胞基因表达数据中,识别出不同的细胞分化状态及其基因的表达模式。简而言之,CSMF是一个用于挖掘不同条件下相互关联生物学数据中,共有和特异生物模式的有效工具。
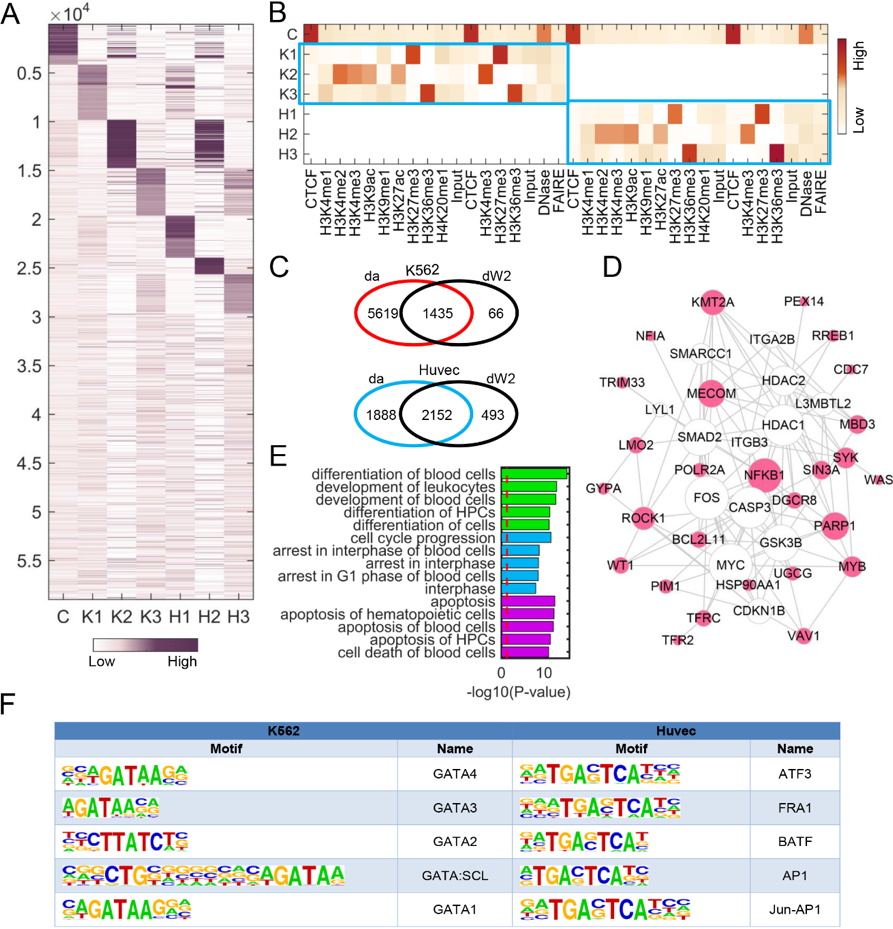
图(二)CSMF识别K562和Huvec细胞系中组蛋白修饰的组合模式。 该研究于当地时间6月8日在线发表在国际学术期刊《Nucleic Acids Research》上,题目为“Learning common and specific patterns from data of multiple interrelated biological scenarios with matrix factorization”。张世华课题组的博士生张丽华为该工作的第一作者。该工作受到中国科学院先导专项、国家自然科学基金、中国科学院前沿重点研究项目等的支持。 |